Scleroderma
Medically reviewed by Kimberly (Showalter) Lakin, MD, MS
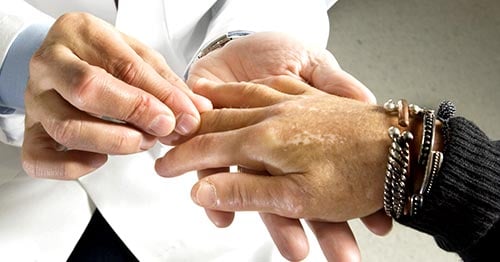
Thickened, hardened, or tightened skin are common symptoms of scleroderma. (The word "scleroderma" means "hard skin.") But this complex disease has different forms as well as a variety of signs and symptoms.
What is scleroderma?
Scleroderma is an autoimmune condition that can lead to a tightening or hardening of the skin and, in some cases, internal organs. It is a chronic condition in which the immune system mistakenly activates in a way that damages your own body. This manifests as an overproduction of collagen, a protein that is a building block of connective tissues. This overproduction leads to fibrosis – an increase of tissue volume – of the skin or of tissues of the lungs or other organs.
Scleroderma Fast Track
Have you recently been diagnosed with scleroderma? If you are looking for an appointment with a rheumatologist specializing in SSc, contact us today.
What causes scleroderma?
As with most autoimmune diseases, the exact cause is unknown. In many patients it may be multifactorial, arising from a combination of congenital processes and environmental agents.
Some scleroderma-like illnesses have also been associated with environmental exposures, such as an outbreak of scleroderma-like illness in Spain in people who had ingested a toxic rapeseed oil. Other cases of scleroderma-like diseases have developed in people who consumed adulterated food containing L tryptophan (an amino acid that is essential to building our bodily proteins, and which we absorb from food).
In some people, certain scleroderma-like lung diseases have occurred after exposures to certain toxins or chemotherapy agents to treat cancer, such as bleomycin.
What are the different types of scleroderma?
There are two basic types of scleroderma: Localized scleroderma and systemic scleroderma (systemic sclerosis). Localized scleroderma affects only the skin, while systemic scleroderma can impact the skin as well as the internal organs. Systemic scleroderma is also known as systemic sclerosis (SSc) and can lead to serious or even life-threatening organ damage and complications.
Localized scleroderma
In some patients, scleroderma is localized and purely a skin disorder, at times characterized by isolated patches of thickened, scarred, tight skin. In other case, the skin involvement becomes more widespread, but the disease still does not extend to affect internal organs or cause vascular (blood vessel) problems.
There are three basic subtypes of localized scleroderma:
- morphea: roundish or irregular-shaped hard patches of skin on the face, hands, feet or trunk
- linear scleroderma: lines or bands of thickened skin on the face, arms or legs
- morphea en coup de sabre: thickened skin causing linear depressions on the forehead only
(Find a localized scleroderma specialist at HSS.)
Systemic scleroderma (systemic sclerosis)
Two major forms of systemic sclerosis are recognized: limited systemic sclerosis (also sometimes referred to as CREST syndrome) and diffuse systemic sclerosis. This second form is also known by many other names, including diffuse cutaneous systemic sclerosis (dcSSc), progressive cutaneous systemic scleroderma, and progressive cutaneous systemic sclerosis. (Find a systemic scleroderma specialist at HSS.)
The difference between limited and diffuse systemic sclerosis depends on the extent of involvement of the skin.
In patients with limited disease, the skin involvement mostly affects the hands, possibly the forearms, and lower extremities, but does not extend above the elbows or knees, and does not involve trunk. The face may be involved as well. In patients with diffuse scleroderma, the skin involvement can also affect the upper arms, thighs, chest wall and trunk.
In both of these systemic forms of scleroderma, vascular (blood vessel) problems such ase Raynaud’s phenomenon are present. Gastrointestinal problems can occur in both forms as well.
There is a rarer subtype known as systemic sclerosis sine scleroderma (ssSSc). In this type of scleroderma, people have unaffected, normal skin but have other manifestations of the systemic scleroderma such as lung involvement or vascular issues.
Limited systemic sclerosis (limited scleroderma)
Limited systemic sclerosis is also sometimes referred to as CREST syndrome, which is an acronym of its common features:
- Calcinosis: nodules of calcium that form under the skin.
- Raynaud’s phenomenon: color changes of the fingers and toes in the cold.
- Esophageal dysfunction: improper functioning of the esophagus, which can cause acid reflux (heartburn) and/or trouble swallowing.
- Sclerodactyly: Tightened skin on the hands that can cause fingers to flex (curl) involuntarily, which can also impair movement.
- Telangiectasia ("spider veins"): Dilated or broken blood vessels located near the surface of the skin or mucous membranes.
The term limited scleroderma is preferred to the term CREST, because the CREST acronym does not include all of the possible symptoms that can occur with this condition. For example, pulmonary hypertension (elevated blood pressure in the lung system) can occur in limited scleroderma and can arise later in the course of the disease. Other types of lung involvement or even kidney involvement can occur in patients with limited disease, though those are less commonly found in limited as compared to diffuse scleroderma.
Diffuse systemic sclerosis (diffuse scleroderma)
Patients with diffuse scleroderma are at higher risk for early lung involvement (lung fibrosis), and doctors will screen for this proactively, even in those without lung symptoms. Some patients with diffuse scleroderma are also at higher risk for kidney involvement. This can be dangerous or life-threatening if left unrecognized, but it can be easily screened for by frequent analysis of urine samples and checking blood pressure regularly (several times a week at home), particularly early in the course of the disease.
What are the symptoms of scleroderma?
Symptoms vary but can include:
- Raynaud’s phenomenon (color changes of the fingers and toes in the cold)
- arthritis or joint pain
- thickened or tightened skin on different parts of the body
- acid reflux, heartburn, and other gastrointestinal problems
- muscle inflammation and/or weakness
More serious manifestations (for systemic scleroderma or systemic sclerosis) include:
- interstitial lung disease
- pulmonary hypertension
- breathing difficulties and chest pain
- anemia
- heart problems
- kidney problems
What are the early signs and symptoms of systemic sclerosis?
Often, the first symptom of systemic sclerosis (systemic scleroderma) is Raynaud’s phenomenon. This is when the fingers turn white, red, and/or blue in response to the cold or stress. However, it is important to note that most people who have Raynaud’s phenomenon do not have scleroderma. Up to 15% of certain populations may have the Raynaud’s phenomenon, while less than 0.001% have systemic sclerosis.
Is scleroderma life-threatening?
Mild forms of scleroderma may disrupt quality of life but poses no danger. But systemic scleroderma is the most lethal of all rheumatic conditions due to the way it can affect organ systems. Systemic scleroderma can lead to pulmonary fibrosis (increased tissue in the lung that can disrupt breathing), pulmonary hypertension (high blood pressure in the arteries that carry blood from the heart to the lungs for oxygen) and scleroderma renal crisis, which can cause kidney failure, heart failure, and other deadly conditions.
The risk and outcomes of these possible complications can be improved by careful monitoring with your rheumatologist and other scleroderma specialists.
Who is most at risk for scleroderma?
Like lupus and rheumatoid arthritis, scleroderma affects women more frequently than men. About 80% of patients with systemic scleroderma are women. This rare disease can develop at any age but usually starts to affect adults sometime between their twenties and their fifties. Localized scleroderma may be seen in adults or children alike. In most patients with scleroderma, there is no clear underlying predisposing risk factor.
Is scleroderma genetic? Does it run in families?
There are some genetic issues which may be associated with a slightly higher risk for scleroderma, but in general, scleroderma does not seem to be a strongly genetically determined disease. The overwhelming majority of patients with scleroderma have no family members with the disorder. A very small number of scleroderma patients (about 1.5%) will have an affected first-degree family member (meaning parent, child or sibling.) These families are being studied by researchers to learn more about scleroderma genetics.
Is scleroderma triggered by environmental factors?
A host of environmental exposures have been associated with the development of scleroderma-like illnesses, but this is relevant in only a minority of patients who ultimately develop scleroderma. Smoking seems to be a risk factor for vascular disease associated with scleroderma and with more severe expression of the disease in general. It is not clear, however, whether tobacco or nicotine use increases the risk of developing scleroderma.
Should I see a dermatologist or rheumatologist if I think I have scleroderma?
A diagnosis of scleroderma frequently requires consultation between several different specialists, including a dermatologist and a rheumatologist. A good place to start might be your primary care doctor, who can help refer you to the correct specialists. Scleroderma can cause symptoms that are like those of other chronic, rheumatic diseases, and it can be confused with other conditions that involve the development of thick and itchy skin. A rheumatologist can help distinguish between them.
Because it is such a rare disease, many physicians are not aware of the screening recommendations nor of available treatment options. People with systemic sclerosis, especially, need a rigorous evaluation for cardiac and lung disease, as well as counseling on how to monitor for the more serious signs and symptoms of this condition. If you are concerned about getting a correct diagnosis or if another physician has diagnosed you with systemic scleroderma (systemic sclerosis), it is very important to be seen by a rheumatologist with special expertise in systemic sclerosis. Please visit our Scleroderma Fast Track page to schedule an expedited appointment with an HSS scleroderma specialist.
How is scleroderma diagnosed?
The scleroderma diagnosis is based on a combination of clinical feature (symptoms) certain laboratory findings. A thorough examination and blood tests are needed to determine whether you have scleroderma. Diagnosis can be difficult, because the disease shares many similarities with other autoimmune disorders. It is very important to consult a rheumatologist who has expertise in scleroderma. Please visit our Scleroderma Fast Track page to schedule an expedited appointment with an HSS scleroderma specialist.
Clinical features of scleroderma
The overwhelming majority of patients with scleroderma had Raynaud’s phenomena, a condition where upon cold exposure extremities can become whitish, bluish, or ultimately even reddish and uncomfortable. Raynaud’s phenomena, however, is common in the general population. In patients with Raynaud’s phenomena in the context of scleroderma, other findings are usually present, including changes that can be observed by an experienced clinician looking closely at the nail beds, skin changes which generally define scleroderma (the typical “hardened skin” that one would see in scleroderma), and the presence of internal organ issues.
Blood tests for scleroderma
A rheumatologist will often order a panel of laboratory tests including autoantibodies, which are markers that can help predict one pattern of disease or another.
Typically, the rheumatologist will order an test for ANA (antinuclear antibody, an antibody commonly found in many of the autoimmune diseases), as well as for Scl-70 antibody (positive in some patients with diffuse scleroderma), anticentromere antibody (generally felt to be a marker for limited scleroderma), and an antibody called RNA polymerase III (which seems to be a marker for diffuse scleroderma, often predicts more rapidly progressive skin disease, and possibly higher risk for kidney involvement in whom blood pressures should be monitored with even greater vigilance). There are other less common antibodies associated with scleroderma that your rheumatologist may also test.
A skin biopsy is usually not necessary to make the diagnosis but can be helpful in some cases and often performed in the context of scleroderma clinical trial studies.
Screening for organ involvement (systemic sclerosis)
There is no single algorithm as to how patients with established or even suspected scleroderma should be screened for organ involvement. The most important screening tool is a comprehensive history, which is a discussion between the patient and the physician, as well as a careful physical examination. Further tests that likely will be ordered will include blood tests and a urine analysis to assess basic parameters of health including whether or not anemia is present, the status of renal function, and usually also muscle blood tests to assess for whether there is any muscle inflammation which can occur in these disorders.
Tests of lung and heart function are recommended for most patients including pulmonary function studies (where a patient breathes into a tube and their lung function is assessed), a chest CT scan, and an echocardiogram which can assess both the heart function, as well as the pressure in the arteries going from the heart to the lung (which helps screen for pulmonary hypertension).
At times, various gastrointestinal (GI) tract tests may be ordered, depending on the patient's symptoms. These tests are all done with an eye towards identifying problems while they are still in an early stage and possibly more easily treatable.
How is scleroderma treated?
There is no cure for this chronic illness, but treatments are available which can help improve pain, various disease manifestations, and improve quality of life. Depending on the severity of the disease, aggressive physical and occupational therapy may be required to prevent disability in some patients.
Several blood-vessel-dilating medications that have been developed have been recognized as being helpful in patients with scleroderma and Raynaud’s phenomenon as well as digital ulcers. Immunosuppressive medications can be used in particular for skin and lung involvement. There have been trials demonstrating that immunosuppressive therapies can be helpful in terms of progression of lung disease in patients with scleroderma and progressive interstitial lung disease. There is also an antifibrotic medication (nintedanib) that is approved to treat progressive lung disease in scleroderma and has been shown in trials to slow the rate of lung function decline in treated patients.
Drug research for scleroderma therapies
A tremendous amount of research is ongoing to find improved treatments and to better understand systemic sclerosis. To learn more about active HSS clinical trials in scleroderma please call 212.774.2048.
There are no drugs that have been unequivocally proven in controlled trials to treat all of the aspects of scleroderma that impact the lives of patients. Encouragingly, there are a number of agents of interest looking at a variety of strategies including immunosuppressive agents such as mycophenolate or cyclophosphamide, antifibrotic therapies with drugs such as tyrosine kinase inhibitors, and even the development of newer biologics which may hold promise in terms of treating the underlying disease.
The scleroderma clinical research community has become much more sophisticated and integrative in our ability to perform the clinical trials necessary to assess these newer medications. Moreover, advances in understanding the basic underlying immune and vascular system abnormalities in scleroderma holds promise for developing newer therapies in the near future. Patients with systemic sclerosis should consider whether they would like to involve themselves in research and be part of finding a cure for this disease.
Can altering my diet help improve scleroderma symptoms?
There is no strong scientific evidence to recommend a specific diet to treat scleroderma, but in general it is good to eat a healthy and balanced diet. However, specific clinical manifestations of scleroderma may improve with dietary modification. Certain foods can trigger gastroesophageal reflux disease (GERD) and avoiding them can improve symptoms. Other issues which may respond to dietary changes include gastrointestinal or esophageal dysmotility, severe weight loss and constipation. Patients with heart or kidney disease as part of scleroderma may require a low salt diet. Sometimes a consultation with a nutritionist is helpful. Be sure to discuss your diet and your concerns with your physician as well.
Scleroderma clinical trials at HSS
Explore Related Patient Stories
View All Patient Stories
Michael Benjamin
New York, NY
Scleroderma

Michelle Grossman
Poughkeepsie, NY
Scleroderma
References
- Bernstein EJ, Assassi S, Castelino FV, Chung L, Correia C, Evnin LB, Frech TM, Gordon JK, Skaug BA, Hant FN, Hummers LK, Sandorfi N, Shah AA, Shanmugam VK, Steen VD, Khanna D. Computed Tomography of the Chest to Screen for Interstitial Lung Disease in Patients With Systemic Sclerosis at Expert Scleroderma Centers in the United States. ACR Open Rheumatol. 2022 Jul;4(7):596-602. doi: 10.1002/acr2.11434. Epub 2022 Apr 23. PMID: 35460213; PMCID: PMC9274361.
- Pelrine ER, Ah-Kioon MD, Zhang M, Barrat FJ, Spiera RF, Gordon JK. Musculoskeletal Involvement in SSc Is Associated with Worse Scores on Short Form-36 and Scleroderma Health Assessment Questionnaire and Lower Tumor Necrosis Factor-α Gene Expression in Peripheral Blood Mononuclear Cells. HSS J. 2016 Oct;12(3):255-260. doi: 10.1007/s11420-016-9515-7. Epub 2016 Jul 22. PMID: 27703420; PMCID: PMC5026664.
- Saketkoo LA, Frech T, Varjú C, Domsic R, Farrell J, Gordon JK, Mihai C, Sandorfi N, Shapiro L, Poole J, Volkmann ER, Lammi M, McAnally K, Alexanderson H, Pettersson H, Hant F, Kuwana M, Shah AA, Smith V, Hsu V, Kowal-Bielecka O, Assassi S, Cutolo M, Kayser C, Shanmugam VK, Vonk MC, Fligelstone K, Baldwin N, Connolly K, Ronnow A, Toth B, Suave M, Farrington S, Bernstein EJ, Crofford LJ, Czirják L, Jensen K, Hinchclif M, Hudson M, Lammi MR, Mansour J, Morgan ND, Mendoza F, Nikpour M, Pauling J, Riemekasten G, Russell AM, Scholand MB, Seigart E, Rodriguez-Reyna TS, Hummers L, Walker U, Steen V. A comprehensive framework for navigating patient care in systemic sclerosis: A global response to the need for improving the practice of diagnostic and preventive strategies in SSc. Best Pract Res Clin Rheumatol. 2021 Sep;35(3):101707. doi: 10.1016/j.berh.2021.101707. Epub 2021 Sep 15. PMID: 34538573; PMCID: PMC8670736.
- Showalter K, Gordon JK. Skin Histology in Systemic Sclerosis: a Relevant Clinical Biomarker. Curr Rheumatol Rep. 2020 Nov 26;23(1):3. doi: 10.1007/s11926-020-00970-z. PMID: 33244633.