Neuropathy Associated with Trisulfated Heparin Disaccharide Antibodies Responsive to IV Immunoglobulin Treatment
From Grand Rounds from HSS: Management of Complex Cases | Volume 8, Issue 3
Case Report
A 56-year-old man with a history of gout and meralgia paresthetica presented with progressive, painful paresthesia since age 53, following a flu-like illness. The paresthesia initially affected his lower legs and then spread to his hands; he described the sensation as burning or crawling in nature. Initial assessment was consistent on nerve conduction studies with a sensorimotor polyneuropathy. Laboratory testing revealed a mildly positive rheumatoid factor and a 0.3g/dL serum immunoglobulin G-κ (IgG-κ) monoclonal gammopathy of undetermined significance (MGUS), though urine immunofixation, vitamin B1, vitamin B12, and thyroid studies were normal, while Lyme antibody testing and transthyretin deoxyribonucleic acid (DNA) testing were negative. An abdominal fat pad biopsy was negative for amyloid deposition. Symptomatic management was attempted with trials of nonsteroidal anti-inflammatory drugs, amitriptyline, gabapentin, pregabalin, and duloxetine, but these were unsuccessful, and he soon required large doses of long-acting and short-acting opioids for pain control.
On evaluation at Hospital for Special Surgery, he described ongoing painful paresthesia, associated with erythema and edema of his legs. These symptoms remained severe despite his intensive opioid regimen, with clear worsening upon heat exposure. He also experienced new-onset photosensitivity, associated with petechial rash (Fig. 1).
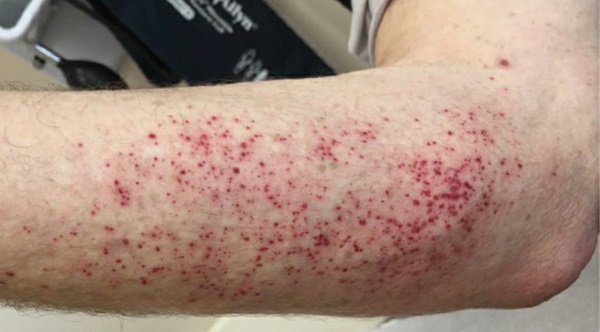
Figure 1: Patient’s petechial rash, which emerged after sun exposure.
Neurologic examination showed normal strength and reflexes, with decreased pain, temperature, and vibration sensation in the distal arms and legs. Electrodiagnostic studies showed normal motor and sensory-evoked responses in the arms and legs, except for mild median neuropathy at the left wrist, and no abnormal spontaneous activity on needle electromyography. Magnetic resonance imaging showed only mild lumbar spine degenerative changes and normal lumbosacral plexus. Skin biopsy showed decreased intraepidermal nerve fiber density at the left calf (2.72 fibers/mm; normal, >3.3) and at the left distal forearm (2.79 fibers/mm; normal, >3). Laboratory testing for antinuclear antibodies, extractable nuclear antibodies, ganglioside, and paraneoplastic antibodies were negative. Immunoglobulin M (IgM) antibodies against IdoA2S-GlcNS-6S, a trisulfated heparin disaccharide (TS-HDS), were then tested and found to be positive (42,000; normal, <10,000).
Partial improvement in symptoms was seen with a trial of moderate-dose corticosteroid therapy. Given the evidence of immune-mediated neuropathy, this was followed by intravenous immune globulin (IVIG), 2g/kg over 4 days, followed by 0.4g/kg every 2 weeks. After starting IVIG therapy, the patient’s symptoms rapidly improved, with less pain and less sensitivity to heat and light. Remarkably, within weeks he no longer required long-acting opioids and could steadily taper the short-acting opioids over 9 months.
Discussion
We report a case of IVIG-responsive, predominantly small-fiber neuropathy associated with IgG-κ MGUS and TS-HDS antibodies. TS-HDS antibodies have previously been associated with immunoglobulin M-κ (IgMκ) MGUS and predominantly small fiber or sensory axonal neuropathy, both in adults [1, 2] and children [3]. As in prior studies, our patient had pure sensory, predominant small-fiber neuropathy. A majority of patients have IgM-κ MGUS; however, our patient had IgG-κ MGUS. Further, the degree of heat sensitivity and the appearance of a petechial rash with sun exposure has not been emphasized previously. Although this is an uncontrolled case study, our patient’s marked decrease in opioid requirement after IVIG therapy began provides a marker of treatment response, along with the decreased rash and photosensitivity. This indicates that IVIG may be a valuable agent in the treatment of this rare condition. Larger-scale, placebo-controlled studies are needed to further validate this observation in other patients.
Posted: 10/1/2019
Authors

Pantelis P. Pavlakis, MD, PhD
References:
- Pestronk A. et al. Sensory neuropathy with monoclonal IgM binding to a trisulfated heparin disaccharide. Muscle Nerve. 2003 Feb;27(2):188-95.
- Pestronk A. et al. Clinical and laboratory features of neuropathies with serum IgM binding to TS-HDS. Muscle Nerve. 2012 Jun;45(6):866-72.
- Kafaie J. et al. Clinical and Laboratory Profiles of Idiopathic Small Fiber Neuropathy in Children: Case Series. J Clin Neuromuscul Dis. 2017 Sep;19(1): 31-37.