Sjogren’s Syndrome-associated Interstitial Lung Disease Treated with Immunosuppression
From Grand Rounds from HSS: Management of Complex Cases | Volume 9, Issue 2
Case Report
A 39-year-old woman with former tobacco use (4.5 pack-years) presented with rapidly progressive interstitial lung disease (ILD). Two years prior, the patient developed subacute exertional dyspnea and cough, for which she received antibiotics without improvement. One year later, she developed diffuse arthralgias (hands, wrists, knees, ankles), xerophthalmia, xerostomia, and worsening dyspnea. Computed tomography (CT) scan of the chest revealed extensive peripheral honeycombing, traction bronchiectasis, and lower lobe predominant ground-glass opacities (Fig. 1). Laboratory testing was notable for positive antinuclear antibody (1:320, speckled) and Sjogren’s syndrome A (anti-SSA) autoantibody. Other laboratory results were unremarkable including rheumatoid factor and serum antibodies to Sjogren’s syndrome B (anti-SSB), anti-Smith, anti-RNP, anti-double-stranded DNA, anti-topoisomerase, anticentromere, RNA polymerase III, myeloperoxidase, serine proteinase 3, anti-Jo-1 (and extended myositis panel), and cyclic citrullinated peptide. Evaluation for infectious etiologies (HIV, tuberculosis, and pulmonary bacterial, viral, and fungal pathogens) was unremarkable.
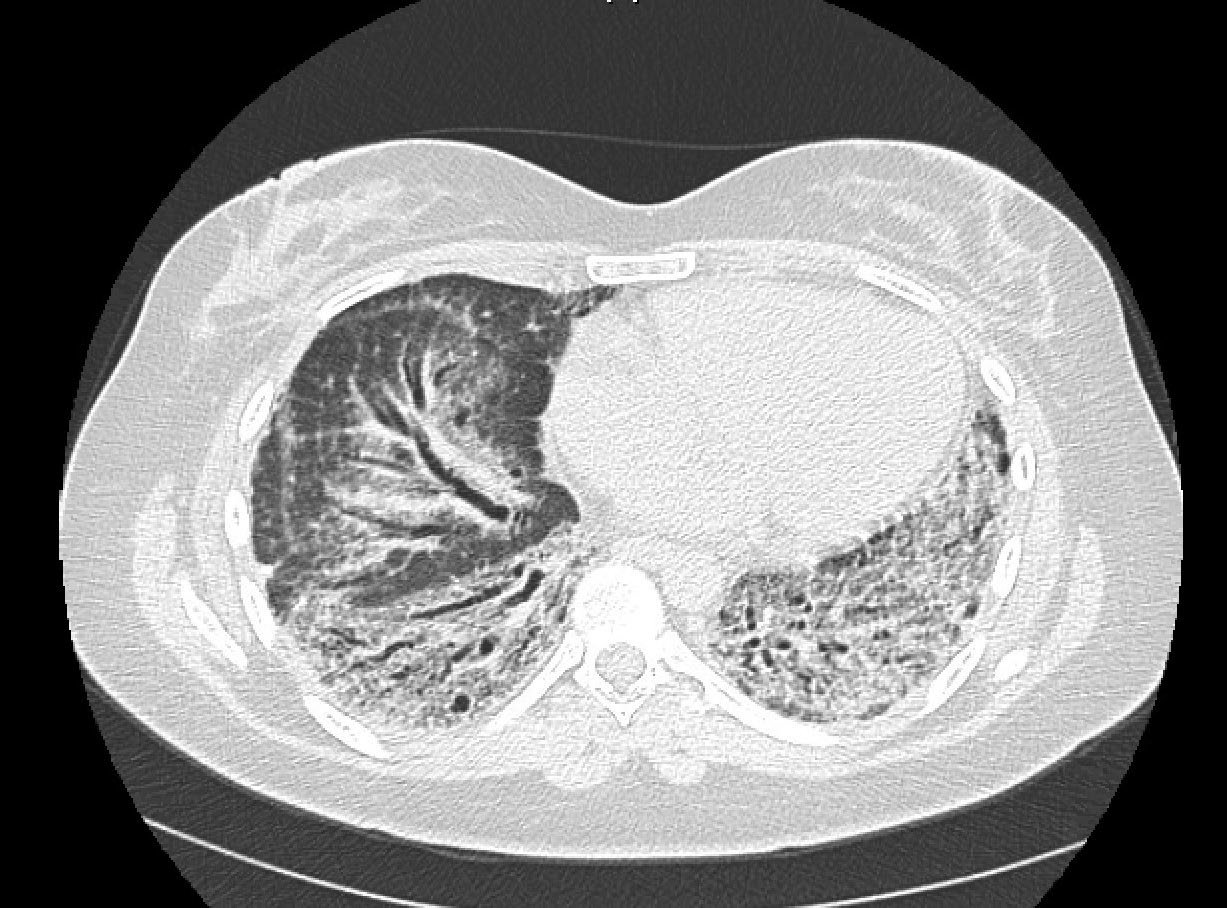
Figure 1: Chest CT scan without contrast demonstrates extensive peripheral areas of honeycombing, areas of traction bronchiectasis, and scattered ground-glass opacities with a lower lobe predominance.
Pulmonary function testing (PFT) demonstrated restrictive lung disease with reduced %-predicted forced vital capacity (FVC) to 32% and normal FEV1/FVC ratio. There was no evidence of pulmonary hypertension on right heart catheterization. A lung biopsy was obtained that demonstrated fibrosing and cellular interstitial pneumonitis with a usual interstitial pneumonia (UIP) pattern. Prednisone (10 mg twice daily) and hydroxychloroquine were initiated without symptomatic improvement. She left the country for 3 months, during which time her shortness of breath slowly worsened. Upon returning, she was hospitalized with acute hypoxemic respiratory failure with FVC decline to 29% predicted and an increasing exertional oxygen requirement (2 L/min to 4 L/min). Repeat infectious work-up was unremarkable and imaging revealed no pulmonary embolism. She was referred both for pulmonary transplantation and rheumatology evaluations.
Additional immunomodulatory medications were added: mycophenolate mofetil (MMF) (increased over 8 weeks to 3 g daily) and rituximab (1 g intravenously for 2 doses, 14 days apart). She also began pulmonary rehabilitation. Subsequently, her joint pain resolved, dyspnea improved, and exertional oxygen requirement decreased to 2 L/min. Her %-predicted FVC improved slightly: 30% and 32% after 6 and 12 months, respectively. Currently, lung transplantation is on hold due to symptomatic improvement.
Discussion
Approximately 10 to 20% of individuals with Sjogren’s syndrome (SS) have clinically relevant pulmonary involvement, defined by respiratory symptoms with PFT and/or chest radiographic abnormalities [1]. Common symptoms include dyspnea, cough, and sputum production. Common histopathologic findings include non-specific interstitial pneumonitis, bronchiolitis, and usual interstitial pneumonia [2]. Factors associated with increased risk of lung disease include male sex, older age, history of tobacco use, hypergammaglobulinemia, lymphopenia, and presence of rheumatoid factor, anti-SSA, or anti-SSB antibodies [1, 3]. Importantly, the presence of ILD is associated with a 4-fold increased risk of death at 10 years [4].
To date, no controlled trial has been conducted in SS-ILD. Prednisone (1 mg/kg) is recommended as initial treatment, and MMF, azathioprine, rituximab, and cyclophosphamide have also been used [1, 5, 6]. MMF was studied in 125 patients with connective tissue disease (CTD)-ILD (5 with SS). Among individuals with UIP, MMF was associated with PFT stability over a median of 2.5 years [6]. Rituximab was evaluated in a retrospective cohort study of SS-ILD (n = 10) [5]. There was no significant 6-month improvement in FVC; however, there was mild improvement in diffusing capacity for carbon monoxide (7%-predicted) and significant improvements in patient reported outcomes. Lung transplantation is considered in patients with advanced pulmonary involvement. A retrospective study of 67,621 patients showed no difference in post-transplantation mortality between those with non-scleroderma CTD-ILD and idiopathic pulmonary fibrosis [7].
Authors



References
- Kreider M, Highland K. Pulmonary involvement in Sjögren syndrome. Semin Respir Crit Care Med. 2014;35(2):255-264.
- Ramos-Casals M, et al. Characterization of systemic disease in primary Sjögren’s syndrome: EULAR-SS Task Force recommendations for articular, cutaneous, pulmonary and renal involvements. Rheumatology (Oxford). 2015;54(12):2230-2238
- Yazisiz V, et al. Lung involvement in patients with primary Sjögren’s syndrome: what are the predictors? Rheumatol Int. 2010;30(10):1317-1324.
- Palm O, et al. Clinical pulmonary involvement in primary Sjögren’s syndrome: prevalence, quality of life and mortality—a retrospective study based on registry data. Rheumatology (Oxford). 2013;52(1):173-179.
- Chen MH, et al. Rituximab therapy in primary Sjögren’s syndrome with interstitial lung disease: a retrospective cohort study. Clin Exp Rheumatol. 2016;34(6):1077-1084.
- Fischer A, et al. Mycophenolate mofetil improves lung function in connective tissue disease-associated interstitial lung disease. J Rheumatol. 2013;40(5):640-646.
- Courtwright AM, et al. Survival and outcomes after lung transplantation for non scleroderma connective tissuerelated interstitial lung disease. J Heart Lung Transplant. 2017;36(7):763-769.
Acknowledgements
We acknowledge Fernando Martinez, MD (Pulmonary and Critical Care Medicine, Weill Cornell Medicine), for his contribution.