Kikuchi–Fujimoto Disease in a 6-Year-Old Boy with a History of Macrophage Activation Syndrome
From Grand Rounds from HSS: Management of Complex Cases | Volume 10, Issue 2
Case Report
A 6-year-old boy with a remote history of massive cervical lymphadenopathy and macrophage activation syndrome (MAS) presented with 1 month of fever and cervical lymphadenopathy suspicious for recurrent MAS. After his birth he had been well until age 2, when he developed fever, rash, lymphadenopathy, elevated inflammatory markers, and cytopenias. Extensive infectious and oncologic workups, including cervical lymph node biopsy, at that time were unrevealing. His symptoms resolved with treatment with steroids and anakinra, which were discontinued within 5 months.
Having been off medication for several years, at age 6 he developed daily fever and massive bilateral cervical lymphadenopathy. Laboratory test results showed elevated erythrocyte sedimentation rate (ESR) (peak: 130 mm/hr), hyperferritinemia (peak: 1228 μg/L), and mild cytopenias (hemoglobin nadir: 8.5 g/dL; platelet nadir: 191 × 109/L; white blood cell count nadir: 2.9 × 109/L). Computed tomography (CT) scanning of the neck showed bilateral cervical lymphadenopathy with a confluent appearance (Fig. 1).
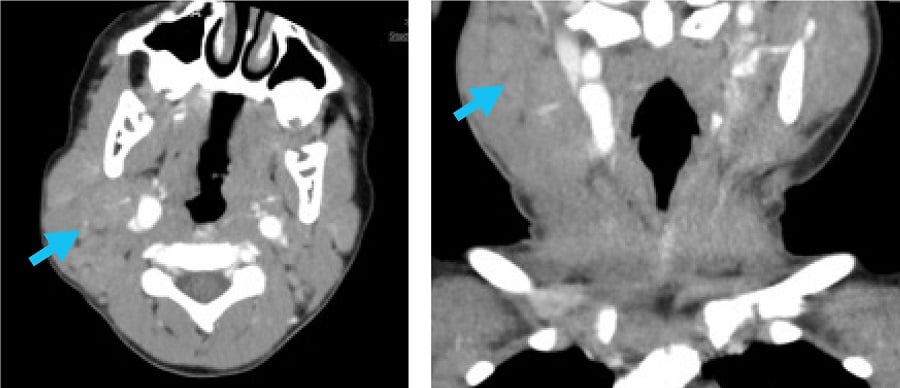 Figure 1: CT (without IV contrast) of the soft tissues of the neck shows right > left enlarged, confluent, bilateral cervical lymph nodes, with arrows pointing to the area of greatest lymphadenopathy.
Figure 1: CT (without IV contrast) of the soft tissues of the neck shows right > left enlarged, confluent, bilateral cervical lymph nodes, with arrows pointing to the area of greatest lymphadenopathy.
Infectious workup was again unrevealing, and repeat excisional cervical node biopsy was performed. The pathology findings showed areas of necrosis with surrounding CD123+ plasmacytoid dendritic cells and mixed cellular infiltrates, consistent with Kikuchi–Fujimoto disease (Figs. 2, 3, 4).
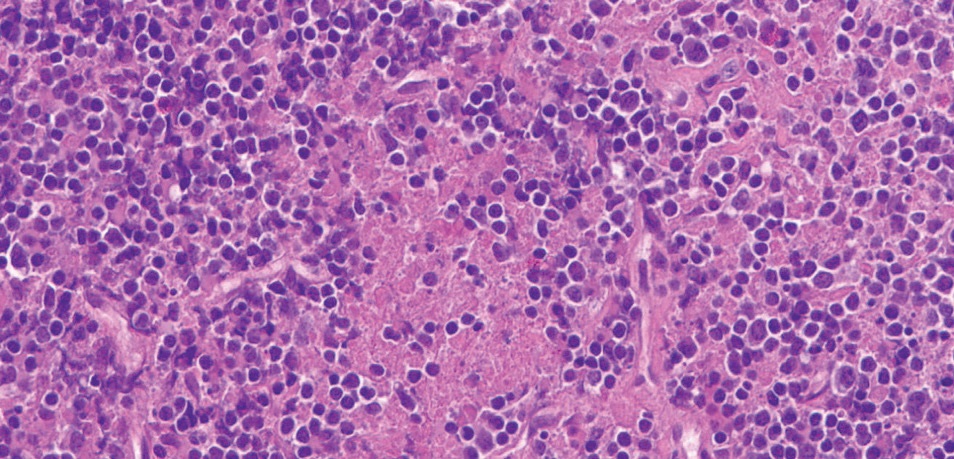
Figure 2: High power, hematoxylin-eosin (H&E) stain shows abundant apoptotic debris and mixed cellular infiltrate comprised of histiocytes, immunoblasts, and small lymphocytes. Neutrophils and plasma cells are essentially absent.
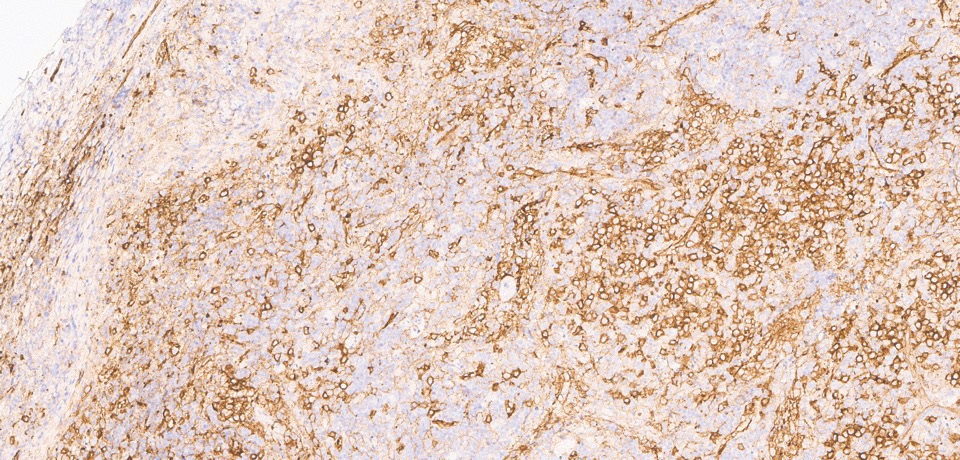
Figure 3: CD123 stain shows many plasmacytoid dendritic cells, predominantly around areas of necrosis.
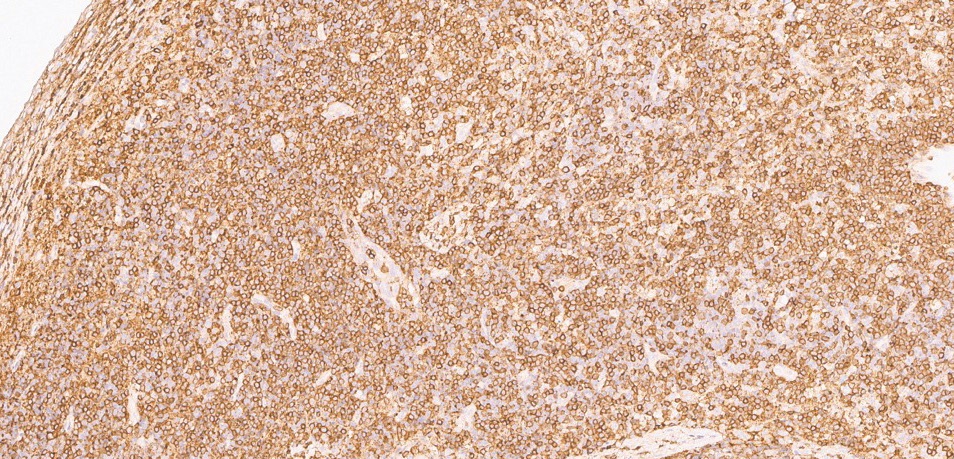
Figure 4: CD3 stain shows abundant T lymphocytes in the infiltrate.
Review of his initial biopsy performed at age 2 showed similar mixed cellular infiltrate with high staining for CD123+ plasmacytoid dendritic cells but did not show the necrosis characteristic of Kikuchi–Fujimoto disease. It was determined that his current presentation was likely a recurrence of his earlier illness, most consistent with recurrent Kikuchi–Fujimoto disease complicated by MAS. Genetic testing for underlying autoinflammatory disease or immunodeficiency was unrevealing.
He was treated initially with 2 mg/kg prednisone daily and 100 mg anakinra daily for 3 days, with rapid resolution of fever and stabilization of cytopenias. He was discharged on a slow steroid taper, but his fever returned and daily anakinra was re-initiated and titrated up to 100 mg BID. He subsequently remained afebrile; all laboratory parameters normalized and lymphadenopathy remitted, allowing for gradual discontinuation of steroids. After 2 months, treatment with hydroxychloroquine was begun for recurrent Kikuchi–Fujimoto disease. After an additional 6 months he remains well and is tolerating a gradual taper of anakinra.
Discussion
Kikuchi–Fujimoto disease is seen increasingly in children, who often do not fit the classical disease presentation. Among children there is predominance in boys (1.4:1 male to female) compared to a predominance in women among adults. Additionally, children with Kikuchi–Fujimoto disease are more likely to present with fever and leukopenia and less likely to have a positive antinuclear antibody test result than adults with the disease [1].
It is hypothesized that Kikuchi–Fujimoto disease is driven by a cytotoxic T-cell mediated inflammatory response to a viral trigger in genetically predisposed individuals. About 3% of patients later develop systemic autoimmune disease, which may be triggered by immune system exposure to autoantigens via the apoptotic debris created in affected lymph nodes [2]. Diagnosis is based on excisional lymph node biopsy, which shows areas of necrosis surrounded by histiocytes and plasmacytoid dendritic cells.
Kikuchi–Fujimoto disease is widely believed to be a benign and self-limited process. However, retrospective studies have shown that up to 30% of hospitalized patients with this diagnosis may develop MAS, often requiring immunosuppressive therapy [3,4].
Kikuchi–Fujimoto disease in children is often recurrent (10%-42% of cases), with up to 3 recurrences in a single patient [5]. Treatment consists of supportive care for mild disease and glucocorticoids for severe or persistent disease, with no treatment trials available. Hydroxychloroquine has been suggested for use in severe or recurrent disease in children [6].
Authors

References
- Kim TY, Ha KS, Kim Y, Lee J, Lee K, Lee J. Characteristics of Kikuchi-Fujimoto disease in children compared with adults. Eur J Pediatr. 2014;173(1):111–116.
- Ogata S, Bando Y, Saito N, Katsuoka K, Ishii M. Kikuchi-Fujimoto disease developed into autoimmune disease: a report of two cases. Mod Rheumatol. 2010;20(3):301–305.
- Ahn SS, Lee B, Kim D, et al. Evaluation of macrophage activation syndrome in hospitalised patients with Kikuchi-Fujimoto disease based on the 2016 EULAR/ACR/PRINTO classification criteria. PLoS One. 2019;14(7):e0219970.
- Duan W, Xiao ZH, Yang LG, Luo HY. Kikuchi’s disease with hemophagocytic lymphohistiocytosis: a case report and literature review. Medicine (Baltimore). 2020;99(51):e23500.
- Selvanathan SN, Suhumaran S, Sahu VK, Chong CY, Tan NW, Thoon KC. Kikuchi-Fujimoto disease in children. J Paediatr Child Health. 2020;56(3):389–393.
- Lin YC, Huang HH, Nong BR, Liu PY, Chen YY, Huang YF, Chiou YH, Lee HS. Pediatric Kikuchi-Fujimoto disease: a clinicopathologic study and the therapeutic effects of hydroxychloroquine. J Microbiol Immunol Infect. 2019;52(3):395–401.