Hereditary Fibrosing Poikiloderma with Tendon Contractures, Myopathy, and Pulmonary Fibrosis (POIKTMP) in a 4-Year-Old with a History of Anti-Synthetase Syndrome
From Grand Rounds from HSS: Management of Complex Cases | Volume 11, Issue 3
Case Report
After relocating from another country, a 4-year-old girl with a previous diagnosis of anti-synthetase syndrome presented for rheumatology care. She had developed an intermittent facial rash at 2 months of age (Fig. 1), and at 4 years of age, a routine laboratory workup demonstrated elevated liver enzymes. Transaminitis was further evaluated for muscle and hepatic etiologies, including a liver biopsy that showed “possible overgrowth and low number of bile ducts,” reportedly abnormal electromyography, and normal lower extremity magnetic resonance imaging (MRI) and lung imaging. Anti-Jo-1 antibody testing came back positive, and a diagnosis of anti-synthetase syndrome was made. She was treated with oral glucocorticoids, mycophenolate mofetil, and ursodiol.
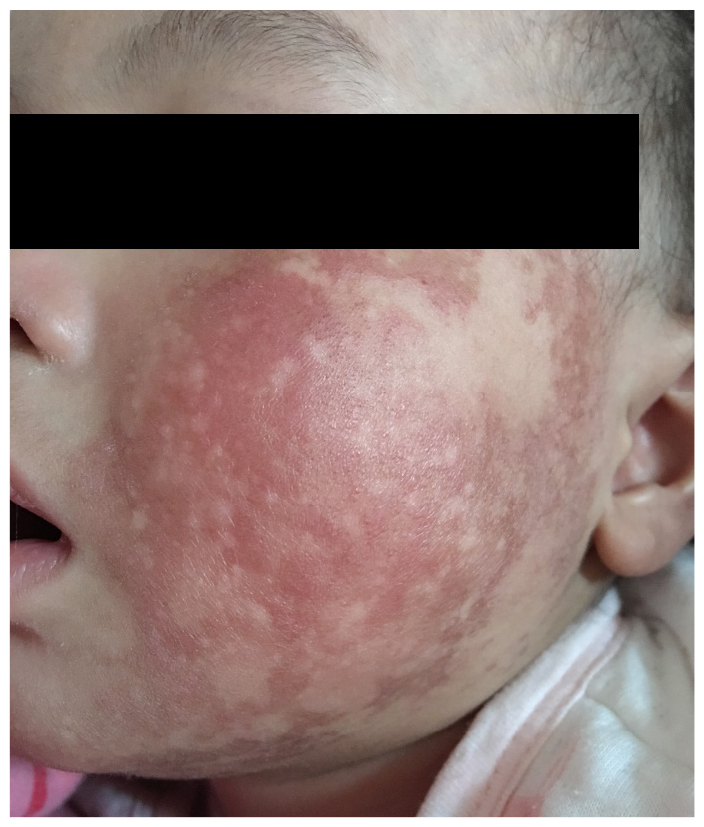
Figure 1: At 2 months of age, the patient had shown signs of poikiloderma, including erythema of the cheeks and face, skin atrophy, telangiectasias, and mottled pigmentation.
After her family’s move to the United States, the girl had normal muscle strength, inflammatory markers, and muscle enzymes. Alanine aminotransferase and aspartate aminotransferase were both elevated, at 304 and 101 units/L, respectively. A myositis panel, including anti-Jo-1 antibodies, was negative, and there was no evidence of myositis on thigh MRI. An ophthalmology consultation found glaucoma secondary to chronic glucocorticoid use; a dermatology consultation noted extremity skin lesions (Fig. 2) and mottled dyschromia of the face and chest (Fig. 3) that was described as poikiloderma (skin atrophy with pigmentation changes and telangiectasias). During a subsequent visit, the girl’s mother was noted to have similar skin findings, prompting a genetics evaluation. Whole exome sequencing showed a mutation of the FAM111B gene, which led to a diagnosis of hereditary fibrosing poikiloderma with tendon contracture, myopathy, and pulmonary fibrosis (POIKTMP).
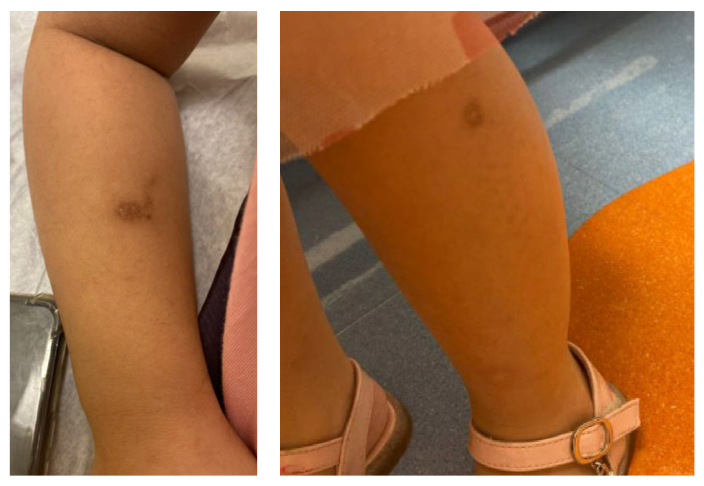
Figure 2: At 4 years of age, the patient developed hemorrhagic bullae on extremities.
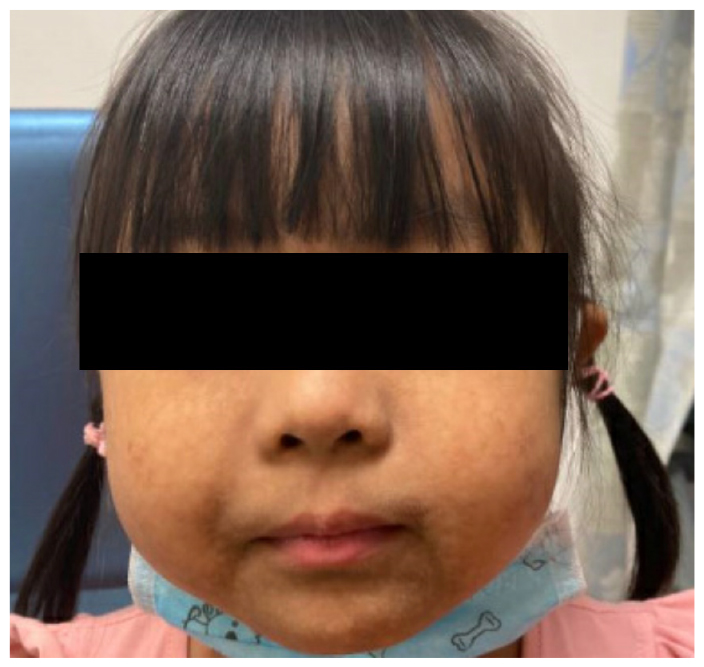
Figure 3: Poikiloderma with mottled hypo- and hyperpigmentation of the patient’s face.
The patient is being treated with ursodiol and sun protection. She has had no other organ system involvement, and her liver enzymes have normalized.
Discussion
POIKTMP is an extremely rare autosomal dominant condition (only about 30 cases have been reported) affecting various organs; a mutation in the FAM111B gene encodes a trypsin-like cysteine/serine peptidase [1]. Skin manifestations often present before 6 months of age, including photosensitive poikiloderma, facial telangiectasias, nonscarring alopecia, mottled pigmentation, bullae, and eczematous lesions [2]. Skin biopsy may demonstrate interface dermatitis or dermal sclerosis [3]. Muscle and tendon contractures are common, although our patient did not develop them, and myopathy can be seen with varying severity. Liver disease may present with transaminitis, hepatomegaly, and lymphocytic ductulitis on biopsy. Lung involvement can progress to dyspnea, pulmonary fibrosis, and restrictive lung impairment, the main cause of death in patients with POIKTMP [4]. Although POIKTMP cannot be cured, the goal remains to manage organ manifestations. Sun protection and close monitoring of organ function are recommended.
Authors
Jheel Pandya, MD
Rheumatology Fellow
Hospital for Special Surgery

Attending Physician, Hospital for Special Surgery
Assistant Professor of Pediatrics, Weill Cornell Medical College
References
- Mercier S, Küry S, Barbarot S. Hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis. 2016 Oct 13 [Updated 2021 Sep 9]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews®. University of Washington, Seattle; 1993-2022. Available from: https://www.ncbi.nlm.nih.gov/books/NBK390610/
- Mercier S, Küry S, Salort-Campana E, et al. Expanding the clinical spectrum of hereditary fibrosing poikiloderma with tendon contractures, myopathy and pulmonary fibrosis due to FAM111B mutations. Orphanet J Rare Dis. 2015;10:135. doi: 10.1186/s13023-015-0352-4
- Dokic Y, Albahrani Y, Phung Tet al. Hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis: Hepatic disease in a child with a novel pathogenic variant of FAM111B. JAAD Case Rep. 2020;6(12):1217-1220. doi: 10.1016/j.jdcr.2020.09.025
- Mercier S, Küry S, Shaboodien G, et al. Mutations in FAM111B cause hereditary fibrosing poikiloderma with tendon contracture, myopathy, and pulmonary fibrosis. Am J Hum Genet. 2013;93(6):1100–1107. doi: 10.1016/j. ajhg.2013.10.013