Erdheim–Chester Disease in a 65-Year-Old Woman Presenting with Retroperitoneal Soft-Tissue Infiltration
From Grand Rounds from HSS: Management of Complex Cases | Volume 12, Issue 3
Case Report
A previously healthy 65-year-old woman presented with acute onset of shortness of breath and chest pain. In addition to conversational dyspnea and distant heart sounds, the patient was noted to have xanthelasmas and light brown macules over the legs, forearms, and back (Fig. 1). Initial workup revealed a large pericardial effusion with tamponade physiology; pericardiocentesis revealed exudative fluid.
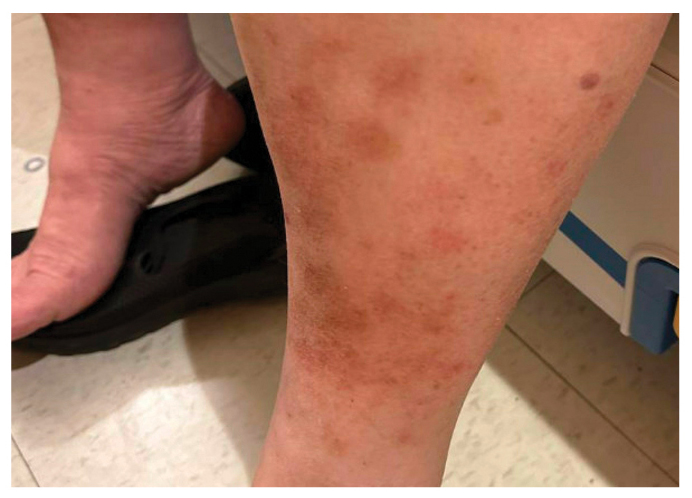
Figure 1: Light brown macules over the legs.
Laboratory workup showed leukocytosis with white blood cell count of 18,000 cell/mm3 and elevated C-reactive protein of 14 mg/dL. Cardiac magnetic resonance imaging (MRI) suggested the presence of an inflammatory process marked by pericardial thickening, myocardial edema, and diffuse late gadolinium enhancement. Cardiac computed tomography (CT) showed non-calcific single-vessel obstructive disease involving the right coronary artery (Fig. 2). A CT scan of the chest, abdomen, and pelvis showed diffuse soft-tissue thickening surrounding the aorta and superior mesenteric artery, as well as the adrenals and the perirenal and retroperitoneal spaces.
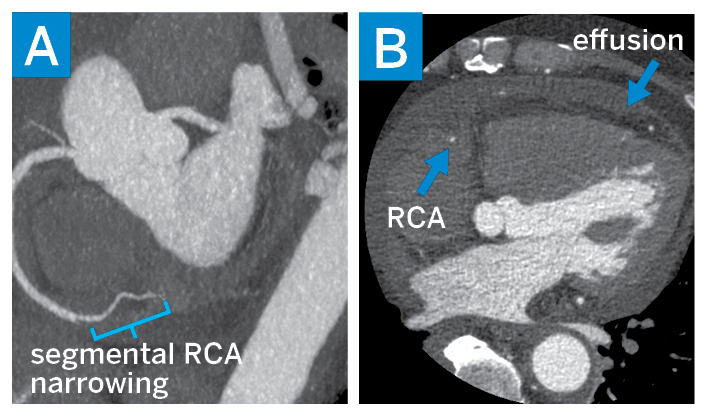
Figure 2: Cardiac CT: (A) a maximum-intensity projection view of the right coronary artery (RCA) showing a narrowing of the mid-RCA and (B) a short-axis view showing a stenotic RCA and pericardial effusion.
These imaging findings raised the concern for vasculitis and an autoimmune infiltrative condition, possibly IgG-4 related disease. Meanwhile, infectious workup, including cultures, respiratory viral panel, and QuantiFERON-TB Gold, was negative. Serologic workup was positive for antinuclear antibodies, at 1:160 in a speckled pattern, but otherwise the autoimmune panel was negative, and IgG4 levels and absolute eosinophil count were within normal range.
A CT-guided adrenal gland biopsy targeting the soft-tissue infiltration that was most avid on positron emission tomography (PET)–CT scanning (Fig. 3) demonstrated an inflammatory infiltrate composed predominantly of histiocytes (highlighted by CD163 immunoreactivity) mixed with lymphocytes, eosinophils, and plasma cells. This raised concern for histiocytosis, particularly Erdheim–Chester disease (ECD). MRI of the legs confirmed the presence of classic sclerotic lesions in the distal tibia and femoral diaphysis. The patient was referred to oncology for further management. A biopsy of the xanthelasma was recommended for mutation testing and was consistent with xanthomatous non-Langerhans histiocytic infiltrate with Touton giant cells in the dermis and extending into skeletal muscle. Mutational analysis of both tumor material and peripheral blood cell-free DNA demonstrated the BRAF V600E mutation. The diagnosis of ECD was confirmed by this constellation of findings, and the patient was started on cobimetinib, a MEK inhibitor. At 2-month follow-up, PET/CT scanning and cardiac MRI showed the pleural and pericardial effusion had completely resolved, with marked decrease in the soft-tissue infiltration.
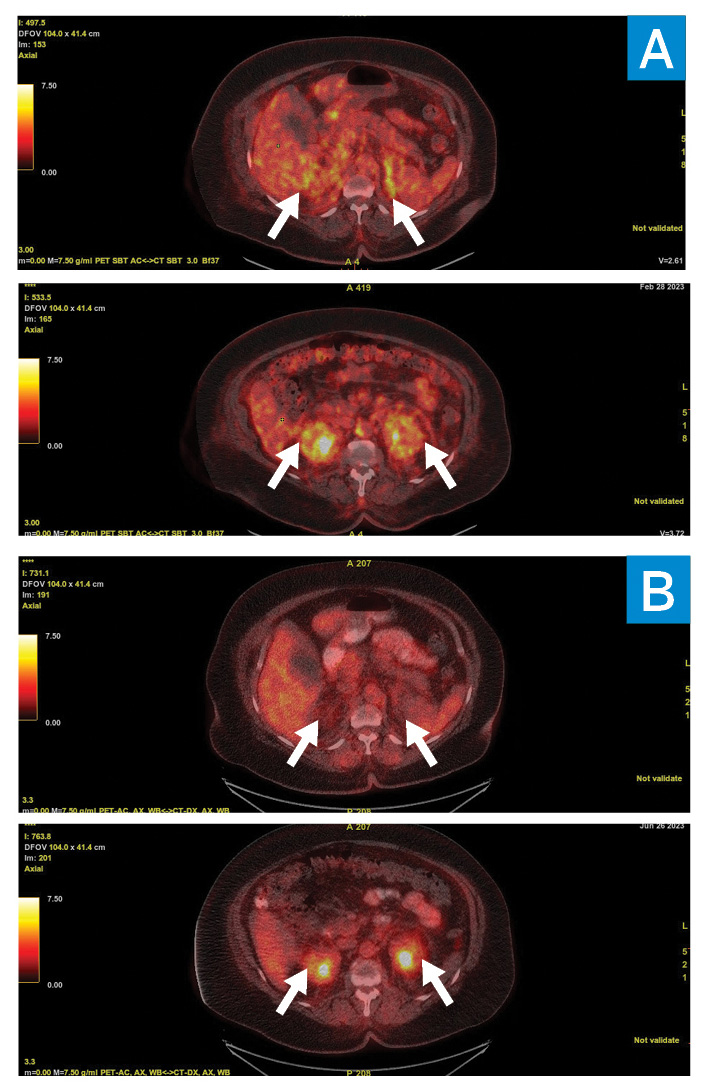
A PET/CT scan showing 18F-fluorodeoxyglucose (FDG)– avidity pre- (A) and post- (B) treatment in the adrenals (top) and perinephric space (bottom).
Discussion
ECD is a rare, multisystem non-Langerhans histiocytosis that was recently recognized as a neoplastic disorder. Disease pathogenesis is characterized by hyperactivation of mitogen-activated protein-kinase (MAPK) signaling leading to histiocytic proliferation and dysregulated chronic inflammation [1]. ECD can affect any age group and sex (with a middle age and male preponderance [2, 3]) and almost any organ; the most common manifestations include long-bone or large-vessel involvement, retroperitoneal fibrosis, or pericardial and myocardial infiltration [4].
Diagnosis requires identifying distinctive histopathological findings. Sclerosing long bone involvement is nearly always present, and histologically lesional tissue demonstrates infiltration of foamy or lipid-laden histiocytes, with admixed or surrounding fibrosis. Touton giant cells are often present [4]. Testing for oncogenic mutations with BRAF V600 sequencing or next-generation sequencing is mandated in all cases to identify targetable alteration for therapy [4]. Therapy should begin at diagnosis, except for patients with minimal symptoms.
As this case highlights, ECD can present in ways that appear consistent with rheumatic diseases, such as vasculitis or retroperitoneal fibrosis, and in the presence of targetable mutations, ECD can be controlled. Early recognition and prompt referral to an academic medical center with expertise in treating ECD are paramount.
Authors
Romy Kallas, MD
Rheumatology Fellow
Hospital for Special Surgery
Eli L. Diamond, MD
Neurologist and Neuro-Oncologist
Memorial Sloan Kettering Cancer Center
References
- Pegoraro F, Papo M, Maniscalco V, Charlotte F, Haroche J, Vaglio A. Erdheim-Chester disease: a rapidly evolving disease model. Leukemia. 2020;34(11):2840-2857.
- Arnaud L, Gorochov G, Charlotte F, et al. Systemic perturbation of cytokine and chemokine networks in Erdheim-Chester disease: a single-center series of 37 patients. Blood. 2011;117(10):2783-2790.
- Estrada-Veras JI, O’Brien KJ, Boyd LC, et al. The clinical spectrum of Erdheim-Chester disease: an observational cohort study. Blood Adv. 2017;1(6):357-366.
- Goyal G, Heaney ML, Collin M, et al. Erdheim-Chester disease: consensus recommendations for evaluation, diagnosis, and treatment in the molecular era. Blood. 2020;135(22):1929-1945.